
Publications
† indicates equal contribution. * indicates corresponding author.
2026
- SCGclust: single-cell graph clustering using graph autoencoders integrating SNVs and CNAs
Potu T.†, Yunfei Hu†, Khan R., Dharani S., Ni J., Zhang L.*, Zhou X. M.*, Mallory X.*
Mathematics, 2026
- ResGAT: sample-efficient graph attention for appendiceal cancer subtypes whole slide image analysis
Z. Lin, H. Tong, Yunfei Hu, X. S. Gui, J. Shen, B. Lee, L. Zhang, D. Moyer, M. Zhou, X. M. Zhou*, K. Votanopoulos
MIDL, 2026
2025

- MaskGraphene: an advanced framework for interpretable joint representation for multi-slice, multi-condition spatial transcriptomics
Yunfei Hu, Lin Z., Xie M., Yuan W., Rao M., Liu Y. H., Shen W., Zhang L.*, Zhou X. M.*
Genome Biology, 2025
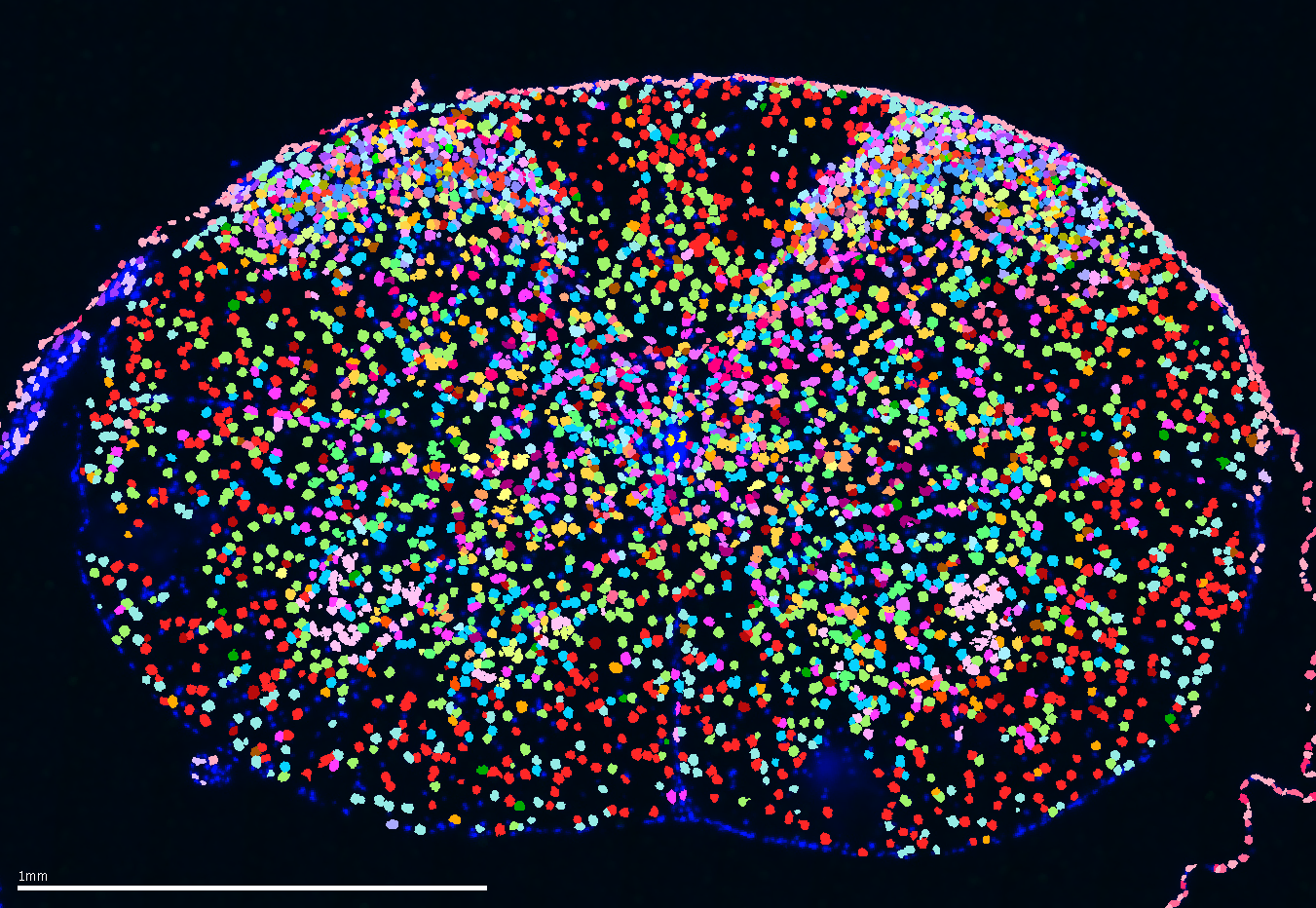
- Spatial transcriptomics reveals organizational properties of mouse spinal cord and alterations in neuropathic pain
Wang Q.†, Yunfei Hu†, Zhu Y., Peng J., Chelliah G., Sobanko M., Sanchez B., Ginty D. D., Zhou X. M., Meltzer S.
Cell Reports, 2025 (under revision)
- An Interpretable Framework for Decoding Transcriptomic Foundation Models
Yunfei Hu†, Wen Z.†, Lin Z., Qin H., Yuan W., Liu Y. H., Klindt D., Zhang L., Carja O., Zhou X. M.
Manuscript under review, 2025
- From features to slice: parameter-cloud modeling of spatial transcriptomics for simulation and 3D interpolatory augmentation
Chen Y., Xie M., Yunfei Hu, Yuan W., Sarkar H., Li B., Zhang L., Zhou X. M.
Preprint, 2025 (manuscript under review)
- BioLACE: unifying spatial geometry and marker priors for cohesive cell-type clustering in spatial transcriptomics
Qin H., Yunfei Hu, Zhu Y., Baek J., Yuan W., Meltzer S., Zhou X. M.
Preprint, 2025 (manuscript under review)
- Interpretable spatial multi-omics data integration and dimension reduction with SpaMV
Liu Y., Ma K., Xu H., Xu K., Yunfei Hu, Lin Z., Lin J., Han B., Li S., Lin Z., Zhou X. M., Zhang L.
Nature Communications, 2025 (under revision)
- CSsingle: a unified tool for robust decomposition of bulk and spatial transcriptomic data across diverse single-cell references
Shen W.*, Yunfei Hu, Liu C., Lei Y., Wong H.-S., Wu S.*, Zhou X. M.*
Nucleic Acids Research, 2025 (under revision)
- CNVeil enables accurate and robust tumor subclone identification and copy number estimation from single-cell DNA sequencing data
Yuan W.†, Luo C.†, Yunfei Hu, Zhang L., Wen Z.-H., Liu Y. H., Mallory X.*, Zhou X. M.*
Preprint, 2025
2024
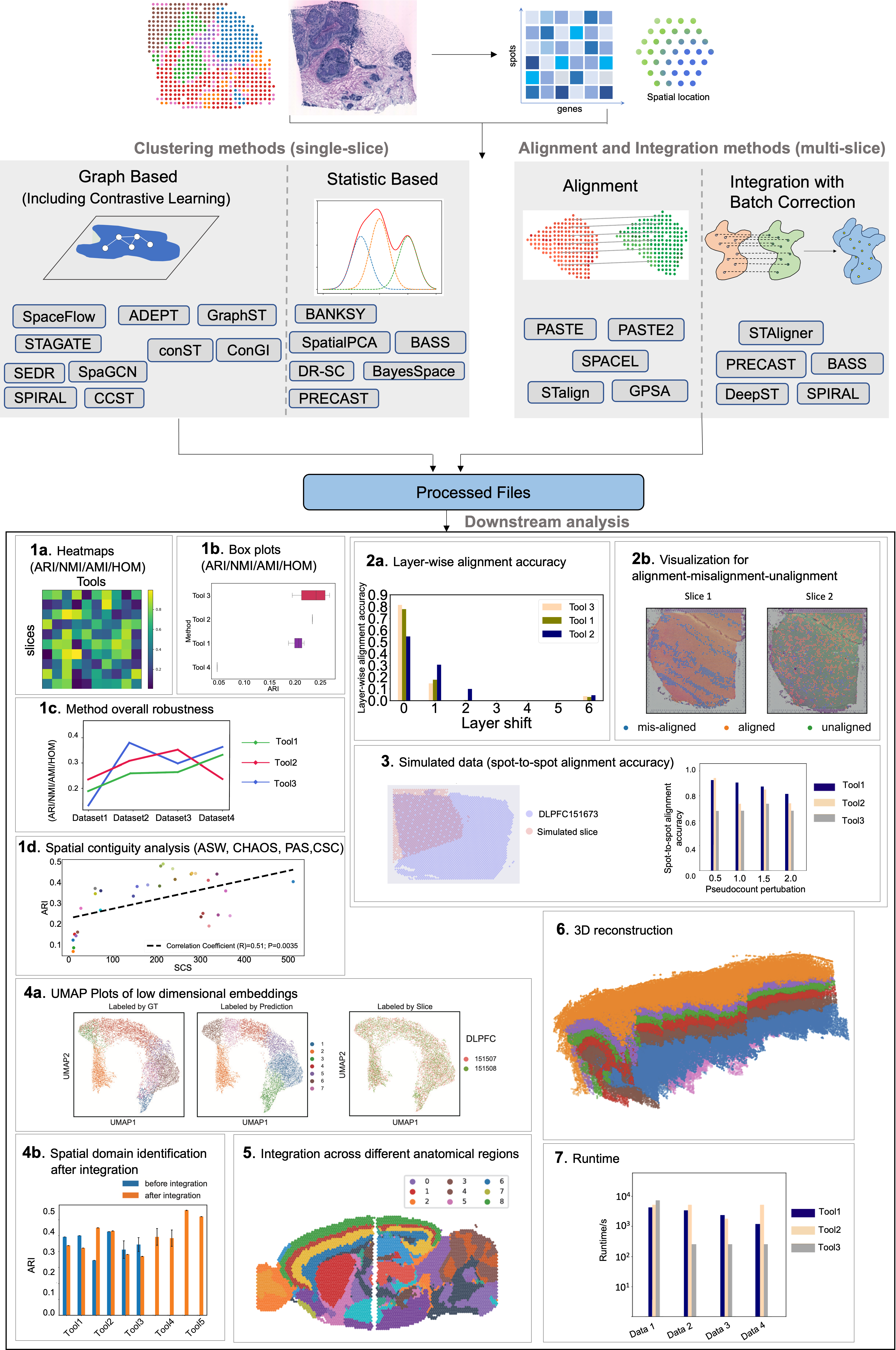
- Benchmarking clustering, alignment, and integration methods for spatial transcriptomics
Yunfei Hu, Xie M., Li Y., Rao M., Shen W., Luo C., Qin H., Baek J., Zhou X. M.
Genome Biology, 2024
2023
- ADEPT: Autoencoder with differentially expressed genes and imputation for robust spatial transcriptomics clustering
Yunfei Hu, Zhao Y., Schunk C. T., Ma Y., Derr T., Zhou X. M.
iScience, 2023
- The hidden patient connections: predicting hormonal therapy medication discontinuation using hypergraph neural networks on clinical communications
Song Q.†, Yunfei Hu†, Ni C., Yin Z.
AMIA Summits on Translational Science Proceedings, 2023
- Haplotyping-assisted diploid assembly and variant detection with linked reads
Yunfei Hu, Yang C., Zhang L., Zhou X.
Methods in Molecular Biology, 2023
2022
- Automated filtering of genome-wide large deletions through an ensemble deep learning framework
Yunfei Hu, Mangal S., Zhang L., Zhou X. M.
Methods, 2022
2021
- Text mining of gene–phenotype associations reveals new phenotypic profiles of autism-associated genes
Li S., Guo Z., Ioffe J. B., Yunfei Hu, Zhen Y., Zhou X. M.
Scientific Reports, 2021